Tools
Primarily, we use statistical approaches to develop methodology for the analysis of human genetics data. We are perhaps more active in development of population genetics methods, but that’s entirely due to the randrom flux of students who have joined the lab. We have plenty of angles for novel method/algorithm development for quantitative genetic analysis (e.g., PRS, Mendelian randomization, fine-mapping)!
Methodolgy Discipline Area:
Population Genetics Focus:
Approaches:
Software Available:
Featured
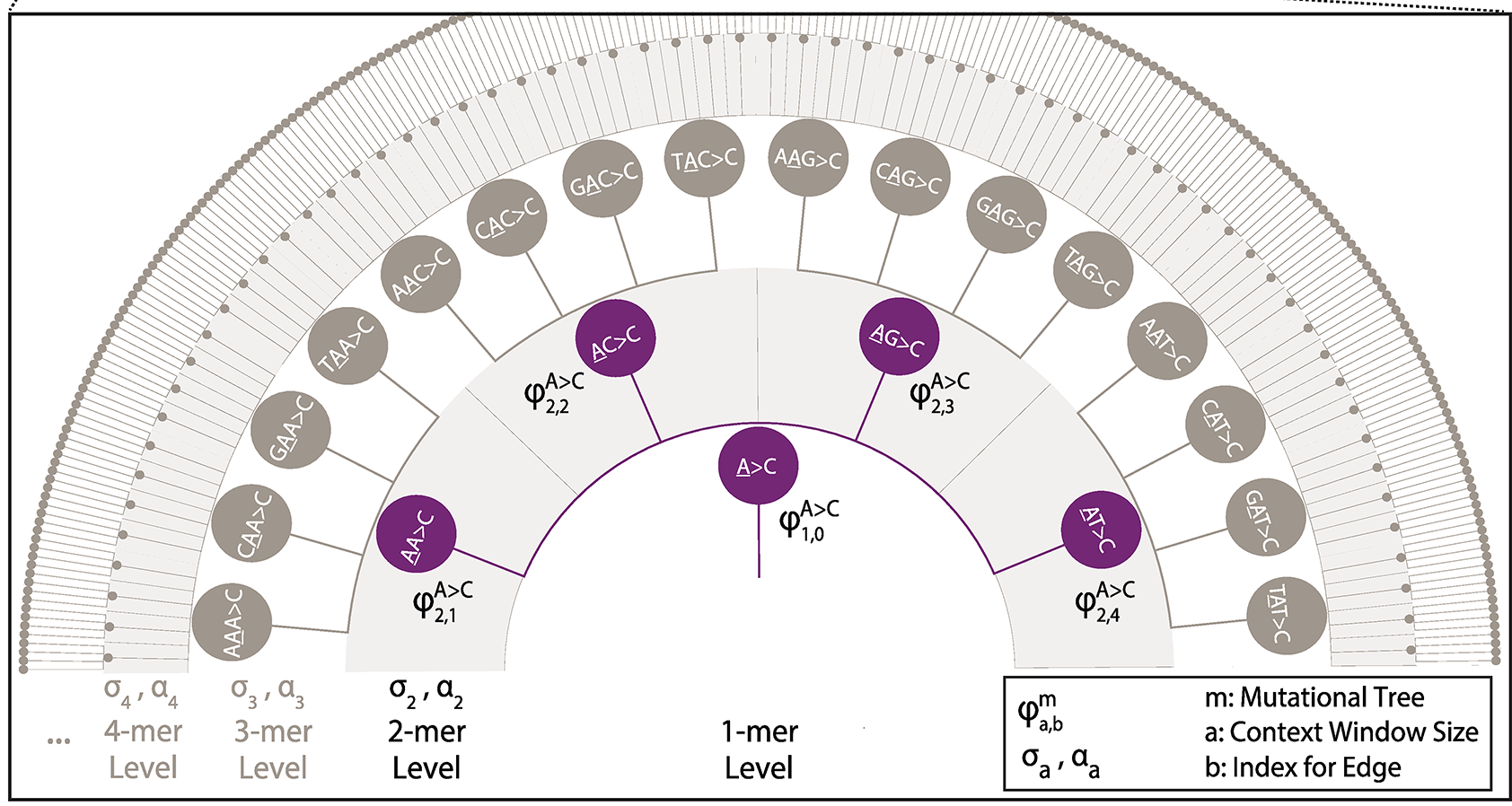
Implementation of a Markov Chain Monte Carlo procedure to estimate hierarchical tree-based sequence context models from variation data sets (polymorphism, substitutions, somatic, de novo, etc.). Introduced by Adams et al (2023).
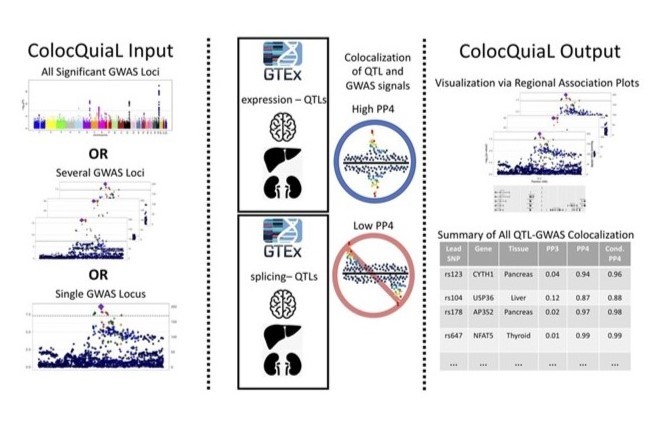
An automated pipeline to parallelize colocalization analyses via COLOC between GWAS and QTL data sets. Described in Chen and Bone et al (2022).
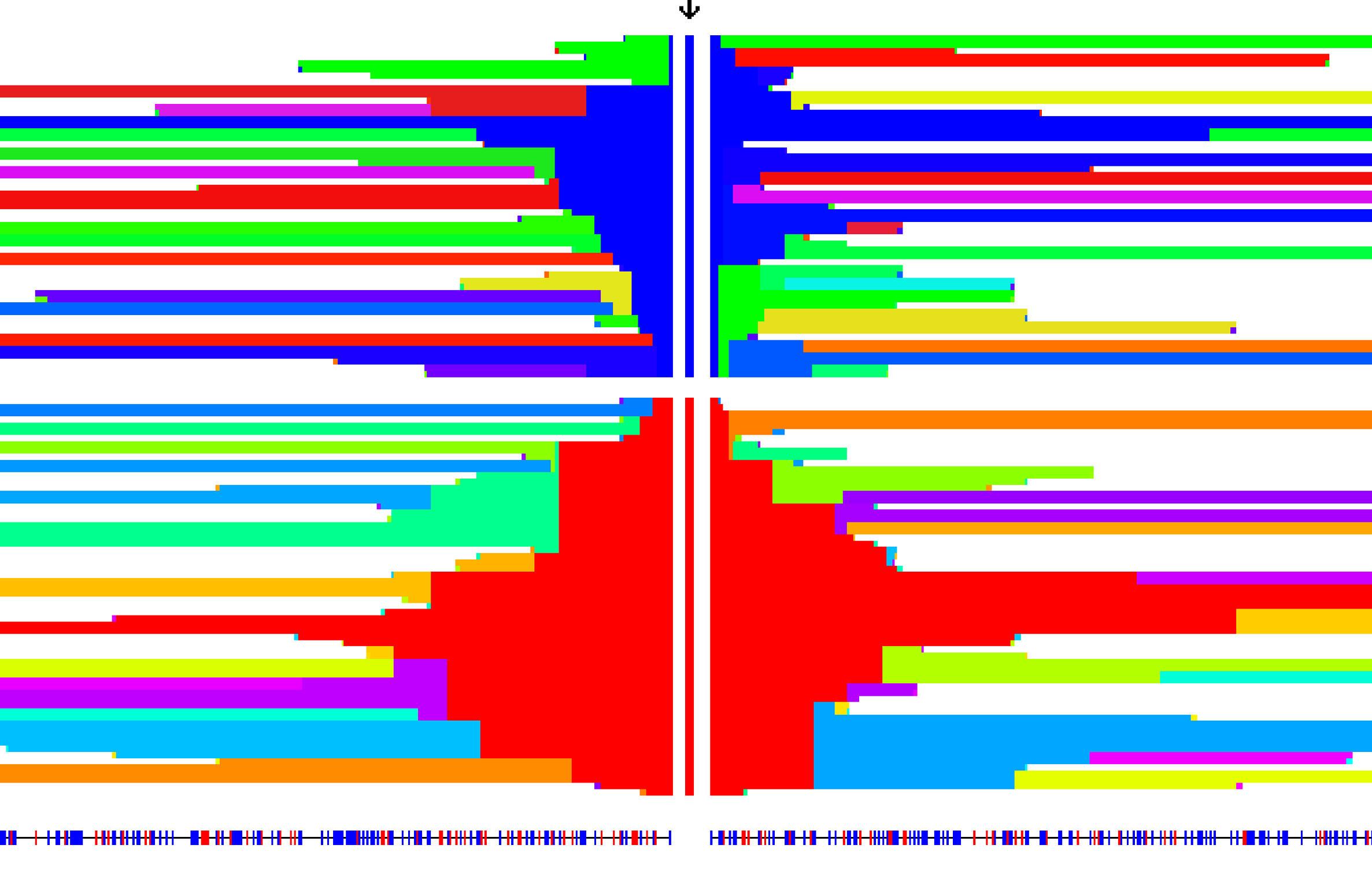
The Intgrated Haplotype Score - A method to detect signals of extended haplotype homozygosity in genomic regions consistent with the action of recent, ongoing selective sweeps using population level data. Introduced in Voight and Kudaravalli et al (2006), adjustments for local, low recombination rates were presented Johnson and Voight (2018).
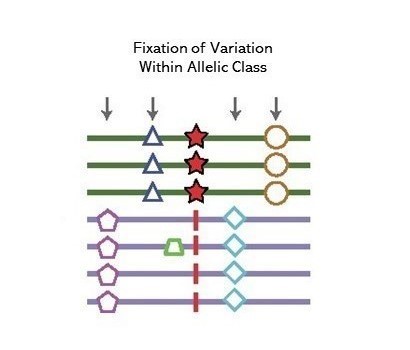
A method to detect signals of an excess number of polymorphisms at near identifical frequency in very close physical proximity, consistent with the action of long-term balancing selection. Introduced in Siewert and Voight (2017), estimates of substitution rates were subsequently incorporated by Siewert and Voight (2020) as the β(2) statistic.
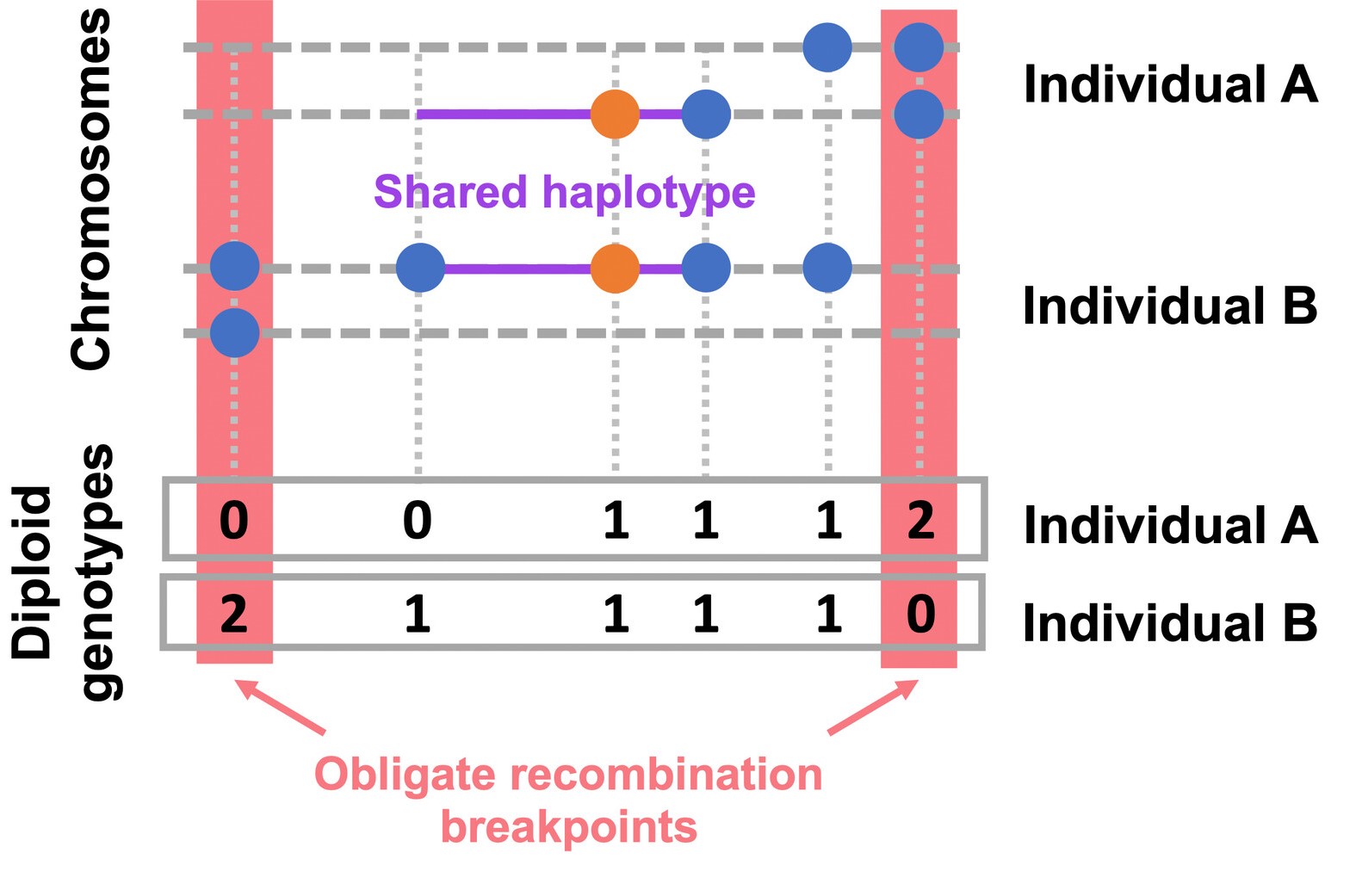
EValuating IBD Consistency via Obligate Recombination Distance - A method designed to detect the rare variants whose alleles are not mutually shared identically by decent, i.e., are IBD-inconsistent which can result from recurrent mutation, gene conversion, or genotype error. This method performs the Gibbs sampler for the Bayesian hierarchical model described by Johnson et al (2022).
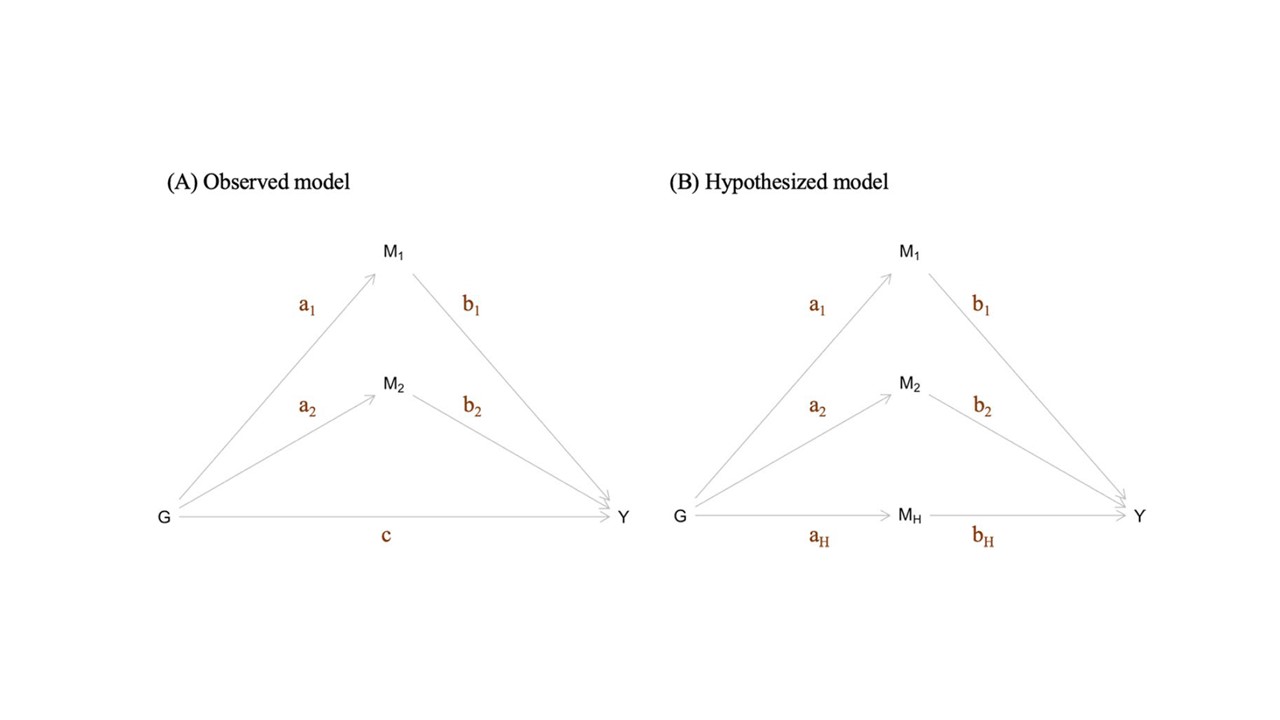
This method is designed, under a simple model, to estimate the effect size of an unknown causal risk factor impacting a collection of SNPs and an outcome trait. Described in Ding et al (2022).

Cross Phenotype Meta-Analysis. Combines p-values across multiple tests into a single test if the distribution of observed p-values are uniformly distributed. Described in Cotsapas et al (2011).

Software designed to simulate causal genetic phenotype models for use with Mendelian randomization studies. Described in Voight (2014).
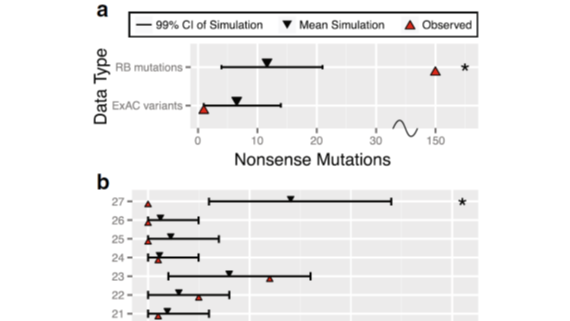
A generalized algorithm to grade the significance of de novo mutational burden in disease probands within a gene based on mutation rate models informed by sequence context. Described in Aggarwala et al (2017).

Presented a statistical framework to evaluate the goodness of fit of competing nested sequence context models. Described in Aggarwala and Voight (2016).
Legacy
Python tool designed to automate the curation of genetic variants selected from the NHGRI GWAS catalog, filtering steps, and causal effect calcualtion for MR. Described in Yin and Voight (2015).